Visualizing the Three-Dimensional Structure of a Molecule
This example shows how to display, inspect and annotate the three-dimensional structure of molecules. This example performs a three-dimensional superposition of the structures of two related proteins.
Introduction
Ubiquitin is a small protein of approximately 76 amino acids, found in all eukaryotic cells and very well conserved among species. Through post-translational modification of a variety of proteins, ubiquitin is involved in many diverse biological processes, including protein degradation, protein trafficking, DNA repair, gene regulation, etc. Because of its ubiquitous presence in cells and its involvement in many fundamental processes, ubiquitin has been the focus of extensive research at the sequence, structural, and functional level.
You can view the three-dimensional structure of ubiquitin by downloading the crystal structure file from the PDB database and then displaying it using the molviewer function. By default, the protein structure is rendered such that each atom is represented by a ball and each bond is represented by a stick. You can change the mode of rendering by selecting display options below the figure. You can also rotate and manipulate the structure by click-dragging the protein or by entering Rasmol commands in the Scripting Console.
In this example, we will explore the structural characteristics of ubiquitin through combinations of Rasmol commands passed to the evalrasmolscript function. However, you can perform the same analysis by using the Molecule Viewer window. The information for the ubiquitin protein is provided in the MAT-file ubilikedata.mat.
load('ubilikedata.mat','ubi')
Alternatively, you can use the getpdb function to retrieve the protein information from the PDB repository and load it into MATLAB®. Note that data in public repositories is frequently curated and updated; therefore the results of this example might be slightly different when you use up-to-date datasets.
ubi = getpdb('1ubi');
h1 = molviewer(ubi);
Warning: MOLVIEWER will be removed in a future release.
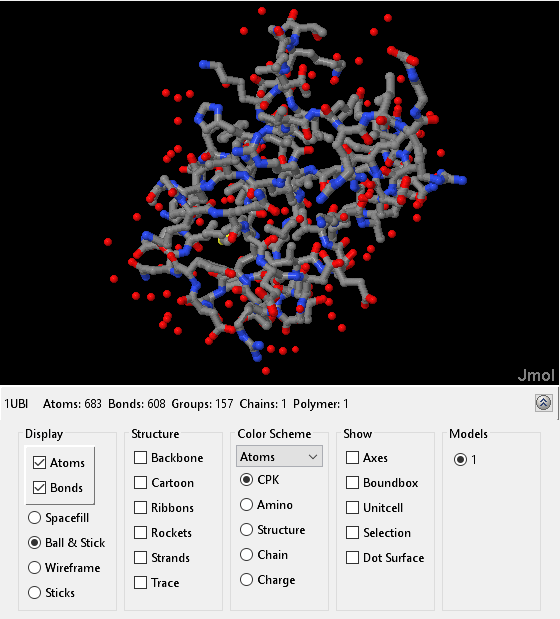
evalrasmolscript(h1, 'select all; wireframe 100; background black;');Warning: EVALRASMOLSCRIPT will be removed in a future release.
Rendering the Molecule
We can look at the ubiquitin fold by using the "cartoon" rendering, which clearly displays the secondary structure elements. We restrict our selection to the protein, since we are not interested in displaying other heterogeneous particles, such as water molecules.
% Display the molecule as cartoon and color the atoms according to their % secondary structure assignment. Then remove other atoms and bonds. evalrasmolscript(h1, ['spacefill off; wireframe off; ' ... 'restrict protein; cartoon on; color structure; ' ... 'center selected;']);
Warning: EVALRASMOLSCRIPT will be removed in a future release.
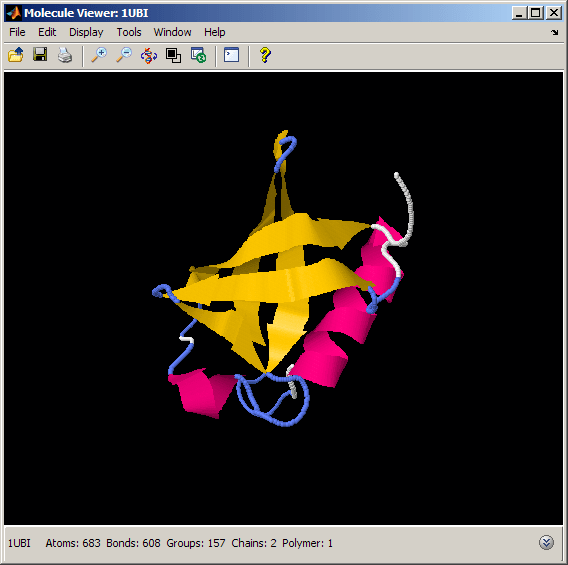
Exploring the Molecule by Spinning and Zooming
The ubiquitin fold consists of five antiparallel beta strands, one alpha helix, a small 3-10 helix, and several turns and loops. The fold resembles a small barrel, with the beta sheet forming one side and the alpha helix forming the other side of the barrel. The bottom part is closed by the 3-10 helix. We can better appreciate the compact, globular fold of ubiquitin by spinning the structure 360 degrees and by zooming in and out using the "move" command.
% Animate the display by making the structure spin and zoom in evalrasmolscript(h1, ['move 0 180 0 40 0 0 0 0 5; ' ... % ... % rotate y by 180, zoom in by 40, time = 5 sec 'move 0 180 0 -40 0 0 0 0 5;']);
Warning: EVALRASMOLSCRIPT will be removed in a future release.
% rotate y by 180, zoom out by 40, time = 5 secEvaluating the Amino Acid Charge Distribution in the Structure
The compactness and high stability of the ubiquitin fold is related to the spatial distribution of hydrophobic and hydrophilic amino acids in the folded state. We can look at the distribution of charged amino acids by selecting positively and negatively charged residues and then by rendering these atoms with different colors (red and blue respectively). We can also render water molecules as white to see their relationship to the charged residues.
evalrasmolscript(h1, ['select protein; color gray; ' ... 'select positive; color red; spacefill 300; ' ... 'select negative; color blue; spacefill 300; ' ... 'select HOH; color white; spacefill 100;']); % water atoms
Warning: EVALRASMOLSCRIPT will be removed in a future release.
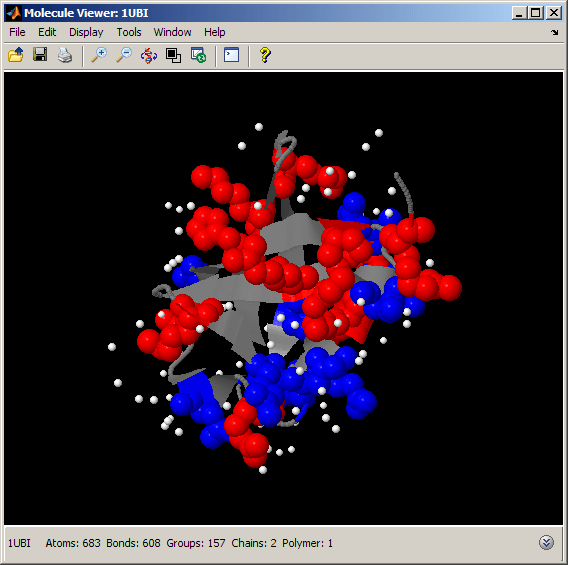
The charged amino acids are located primarily on the surface exposed to the solvent, where they interact with the water molecules. In particular, we notice that the charge distribution is not uniform across the sides of the ubiquitin's barrel. In fact, the side with the alpha helix appears to be more crowded with charged amino acids than the side containing the beta strands.
Exploring the Hydrophobicity Profile of the Structure
We can perform a similar analysis by looking at the spatial distribution of some hydrophobic amino acids, such as Alanine, Isoleucine, Valine, Leucine and Methionine. You can also use the Rasmol label "hydrophobic" to select all hydrophobic residues.
% color hydrophobic amino acids green evalrasmolscript(h1, ['select all; spacefill off; color gray; ' ... 'select Ala or Ile or Val or Leu or Met; ' ... 'color green; wireframe 100;' ... 'move 90 0 0 0 0 0 0 0 1; move 0 -45 0 0 0 0 0 0 1']);
Warning: EVALRASMOLSCRIPT will be removed in a future release.
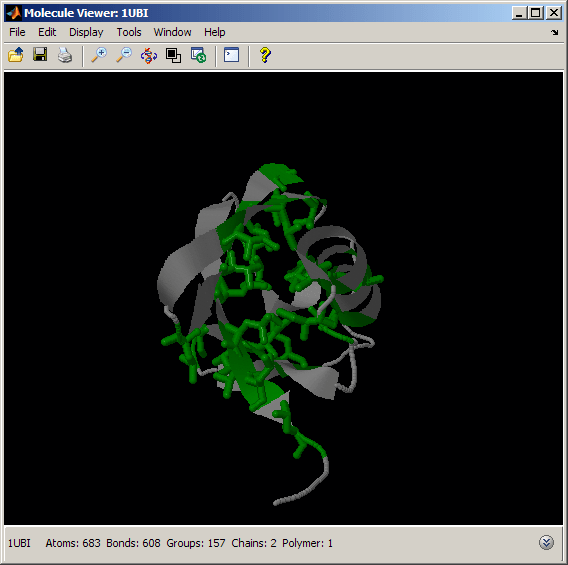
Unlike the charged amino acids above, the hydrophobic amino acids are located primarily in the interior of the barrel. This gives high stability to the ubiquitin fold, since hydrophobic amino acids are shielded from the solvent, making the protein structure compact and tight.
Measuring Atomic Distances
Ubiquitin displays a tight fold with one alpha helix traversing one side of the small barrel. The length of this alpha helix presents some variation among the representatives of the ubiquitin-like protein family. We can determine the actual size of the helix either by double clicking on the relevant atoms or by using MATLAB® and Rasmol commands as follows.
% reset the display to cartoons evalrasmolscript(h1, ['reset; select all; spacefill off; wireframe off; '... 'cartoon on; color structure;']);
Warning: EVALRASMOLSCRIPT will be removed in a future release.
% determine the boundaries of the alpha helix initHelixRes = ubi.Helix(1).initSeqNum % alpha helix starting residue
initHelixRes = 23
endHelixRes = ubi.Helix(1).endSeqNum % alpha helix ending residueendHelixRes = 34
% highlight the starting and ending residues of helix evalrasmolscript(h1, ['select ' num2str(initHelixRes) ' or ' ... num2str(endHelixRes) '; color red; wireframe 100;']);
Warning: EVALRASMOLSCRIPT will be removed in a future release.
% determine atom numbers for starting and ending residues initHelixAtoms = ubi.Model.Atom([ubi.Model.Atom(:).resSeq]==initHelixRes); endHelixAtoms = ubi.Model.Atom([ubi.Model.Atom(:).resSeq]==endHelixRes); initHelix = min([initHelixAtoms.AtomSerNo]); % Helix starting atom endHelix = min([endHelixAtoms.AtomSerNo]); % Helix ending atom evalrasmolscript(h1, ['measure ' num2str(initHelix) ' ' num2str(endHelix) ';']);
Warning: EVALRASMOLSCRIPT will be removed in a future release.
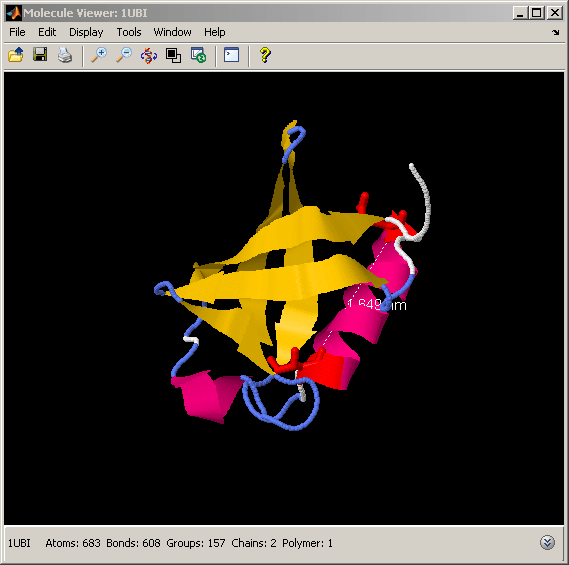
Displaying and Labeling Lysine Residues in Ubiquitin Structure
The process of ubiquitination - the attachment of a ubiquitin molecule to a target protein - is mediated by the formation of an isopeptide bond between the C-terminal 4-residue tail of ubiquitin and a Lysine of the target protein. If the target protein is another ubiquitin, the process is called polyubiquitination. Polyubiquitin chains consisting of at least four ubiquitins are used to tag the target proteins for degradation by the proteasome. All seven Lysines in ubiquitin can be used in the polyubiquitination process, resulting in different chains that alter the target protein in different ways. We can look at the spatial distribution of Lysines on the ubiquitin fold by selecting and labeling the alpha carbons of each Lysine in the structure.
% highlight the Lysine residues in the structure and the C-terminal tail % involved in the isopeptide bond formation evalrasmolscript(h1, ['restrict protein; cartoon off; wireframe off; measure off; ' ... ... % undo previous selection 'backbone 100; color structure; select Lys; wireframe 100; ' ... ... % select Lysines 'select Lys and *.ca; spacefill 300; labels on; ' ... ... % label alpha carbons 'select 72-76; wireframe 100; color cyan; ']);
Warning: EVALRASMOLSCRIPT will be removed in a future release.
% select C-terminal tail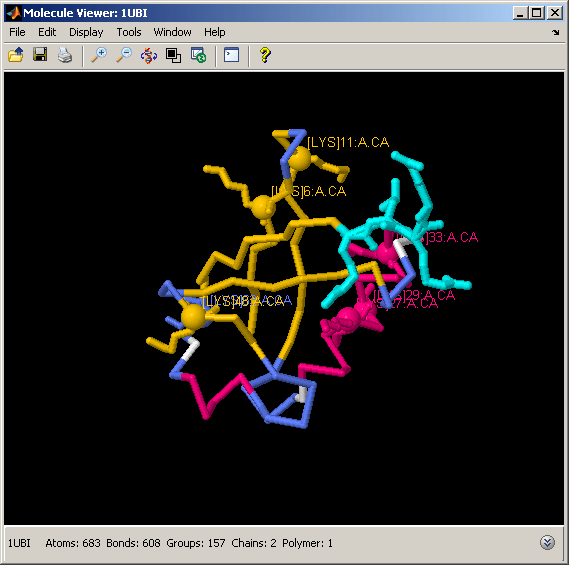
Several studies have shown that different roles are played by polyubiquitins when the molecules are linked together through different Lysines. For example, Lys(11)-, Lys(29)-, and Lys(48)-linked polyubiquitins target proteins for the proteasome (i.e., for degradation). In contrast, Lys(6)- and Lys(63)-linked polyubiquitins are associated with reversible modifications, such as protein trafficking control.
Examining the Isopeptide Bond in Diubiquitin
The crystal structure of a diubiquitin chain consisting of two moieties is represented in the PDB record 1aar. We can view and label an actual isopeptide bond between the C-terminal tail of one ubiquitin (labeled as chain A), and Lys(48) of the other ubiquitin (labeled as chain B).
Retrieve the protein 1aar from PDB or load the data from the MAT-file.
aar = getpdb('1aar');
load('ubilikedata.mat','aar') h2 = molviewer(aar);
Warning: MOLVIEWER will be removed in a future release.

evalrasmolscript(h2, ['restrict protein; color chain; ' ... 'spacefill off; wireframe off; ' ... 'cartoon on; select 76:A, 48:B; spacefill; ' ... ... % isopeptide bond 'select 76:A and *.ca; ' ... % select alpha carbon 'set labeloffset 40 10; label isopeptide bond; ' ... 'move 0 360 0 -20 0 0 0 0 5; ']); % animate
Warning: EVALRASMOLSCRIPT will be removed in a future release.
Aligning Ubiquitin and SUMO Sequences
There is a surprisingly diverse family of ubiquitin-like proteins that display significant structural similarity to ubiquitin. One of these proteins is SUMO (Small Ubiquitin-like MOdifier), a small protein involved in a wide spectrum of post-translational modifications, such as transcriptional regulation, nuclear-cytosolic transport, and protein stability. Similar to ubiquitination, the covalent attachment and detachment of SUMO occur via a cascade of enzymatic actions. Despite the structural and operational similarities between ubiquitin and SUMO, these two proteins display quite limited sequence similarity, as can be seen from their global sequence alignment.
Retrieve the protein SUMO from PDB or load the data from the MAT-file.
aar = getpdb('lwm2');
load('ubilikedata.mat','sumo')
Align the two primary sequences from both compounds.
[score aln] = nwalign(ubi.Sequence.Sequence, sumo.Sequence.Sequence)
score = -3.3333
aln = 3x82 char array
'MQ----I-F-VKTLTGKTITLEVEPSDTIENVKAKIQDKEGIPPDQQRLIFAGKQLEDGRTLSDYNIQKESTLHLVLRLRGG'
' : | : | |::: :::: : :: :::|: | |: | |: ::: | :: ::: |:|: |:: : '
'TENNDHINLKVAGQDGSVVQFKIKRHTPLSKLMKAYCERQGLSMRQIRFRFDGQPINETDTPAQLEMEDEDTID-VFQ-Q--'
Superposing the Structures of Ubiquitin and SUMO
In order to better appreciate the structural similarity between ubiquitin and SUMO, perform a three-dimensional superposition of the two structures. Using the pdbsuperpose function, we compute and apply a linear transformation (translation, reflection, orthogonal rotation, and scaling) such that the atoms of one structure best conform to the atoms of the other structure.
close (h1, h2); % close previous instances of molviewerpdbsuperpose(ubi, sumo, Display=true);
Warning: PDBSUPERPOSE will not accept the DISPLAY name-value argument in a future release.
Warning: MOLVIEWER will be removed in a future release.

Warning: EVALRASMOLSCRIPT will be removed in a future release.
Warning: EVALRASMOLSCRIPT will be removed in a future release.
h3 = findobj('Tag', 'BioinfoMolviewer'); % retrieve handle for molviewer evalrasmolscript(h3, ['select all; zoom 200; center selected']);
Warning: EVALRASMOLSCRIPT will be removed in a future release.
evalrasmolscript(h3, ['select all; cartoons off; ' ... 'select model = 1; strands on; color red; ' ...% ubiquitin 'select model = 2; strands on; color blue;']); % SUMO
Warning: EVALRASMOLSCRIPT will be removed in a future release.
By selecting the appropriate option button in the Models section of the Molecule Viewer window, we can view the ubiquitin structure (Model = 1) and the SUMO-2 structure (Model = 2) separately or we can look at them superposed (Model = All). When both models are actively displayed, the structural similarity between the two folds is striking.
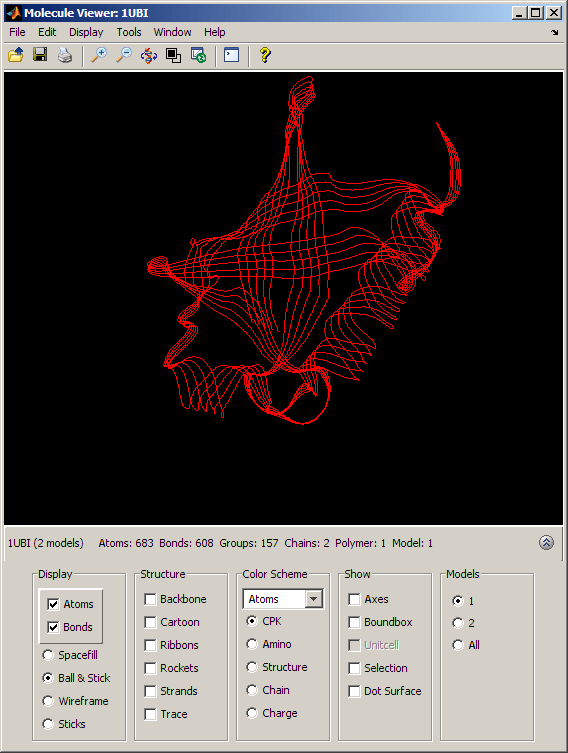
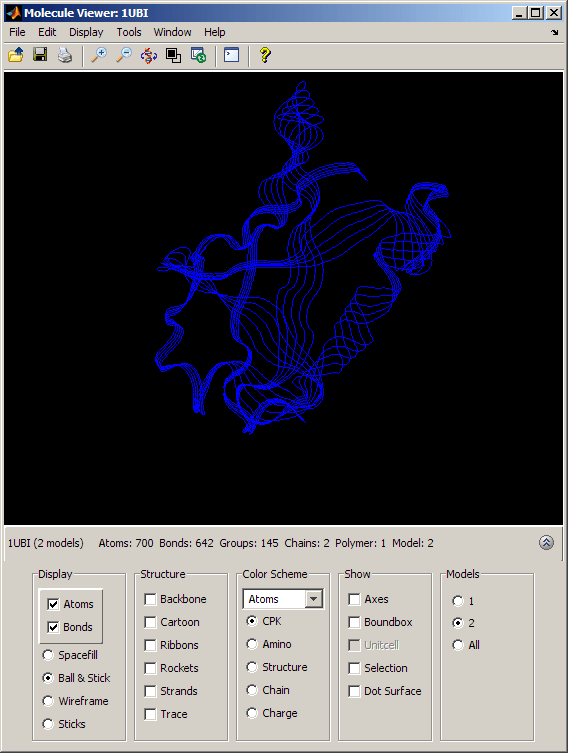
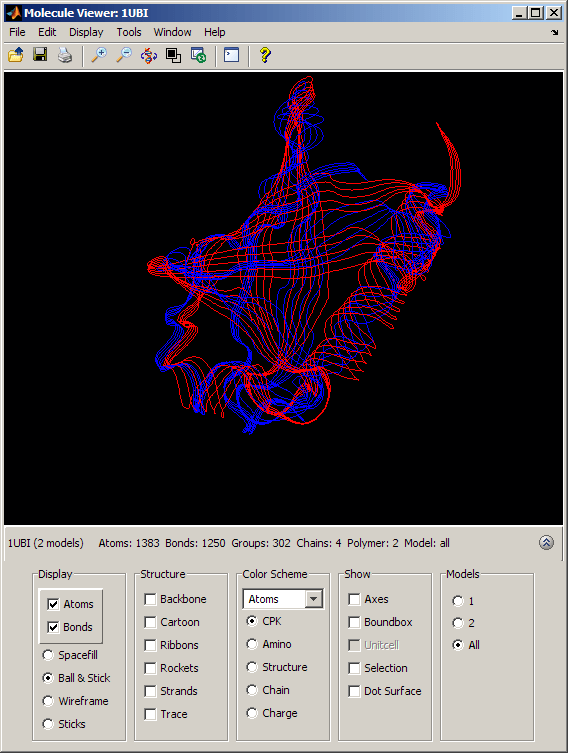
The conservation of the structural fold in the absence of a significant sequence similarity could point to the occurrence of convergent evolution for these two proteins. However, some of the mechanisms in ubiquitination and sumoylation have analogies that are not fold-related and could suggest some deeper, perhaps distant, relationship. More importantly, the fact that the spectrum of functions performed by ubiquitin and SUMO-2 is so widespread, suggests that the high stability and compactness of the ubiquitin-like superfold might be the reason behind its conservation.
close all; You can also select a web site from the following list:
Americas
- América Latina (Español)
- Canada (English)
- United States (English)
Europe
- Belgium (English)
- Denmark (English)
- Deutschland (Deutsch)
- España (Español)
- Finland (English)
- France (Français)
- Ireland (English)
- Italia (Italiano)
- Luxembourg (English)
- Netherlands (English)
- Norway (English)
- Österreich (Deutsch)
- Portugal (English)
- Sweden (English)
- Switzerland
- United Kingdom (English)